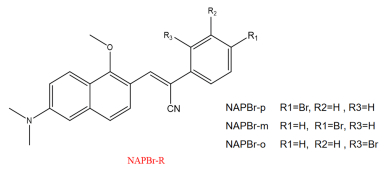
Figure
1.
Structural formulas of the studied compounds.
| Citation: |
Hong-yang Wang, Xiao-fei Wang, Jian-zhong Fan, Yong Zhou. Effect of Substitution Site on Two-Photon Absorption and Excited States Properties of Lipid Droplets Detection NAPBr Dyes: a Theoretical Perspective[J]. Chinese Journal of Chemical Physics , 2022, 35(4): 673-684. DOI: 10.1063/1674-0068/cjcp2007134

|
Lipid droplets are lipid-rich cellular dynamic organelles that regulate the storage and hydrolysis of neutral lipids. Lipid droplets are found to have great quantity in the adipose tissues. Lipid droplets are important to support normal cellular functions, including lipid metabolism, signal transduction, protein degradation, membrane synthesis and transfer, etc. [1, 2]. Recently, lipid droplets have been found to have a close association with inflammatory responses through the synthesis and metabolism of eicosanoids and metabolic disorders, such as obesity, diabetes, inflammatory disorders, cancers and atherosclerosis [3-6]. Therefore, it is important to find a method that can monitor lipid droplets in samples such as living biological tissues effectively and selectively. Compared with other detection methods, the two-photon fluorescence imaging is more conducive to biomedical imaging due to its advantages such as large penetration depth, low tissue auto-fluorescence, self-absorption, and low photo-damage and photo-bleaching [7-9]. Moreover, Tang's group pays more attention to aggregation-induced emission (AIE). They use the two-photon naphthalene-based donor-acceptor NAP AIEgens molecules that introduce a large heterocyclic ring to increase the electron-withdrawing ability of acceptor, and the specific two-photon imaging of living cells and deep tissue lipid droplets are studied [2]. However, the molecular properties and their intrinsic mechanism analysis are not illustrated sufficiently. The restriction of intramolecular rotations (RIR) is the main reason for AIE, it affects the excited state energy consumption process by non-radiative transitions. It weakens fluorescence or even not emits.
In this work, we focuses on the substitution site effect on the molecular geometric structural changes, non-radiative transitions, photophysical properties and internal mechanisms. It deepens the understanding and provides strategies for the next step of our research about the substitution site effect on AIE and two-photon absorption (TPA) properties. Molecules with different substitution sites may have their unique advantages, such as excellent TPA ability or high fluorescence quantum efficiency. It may even provide a reference for excellent dyes as needed in actually selecting. In this work, the relationship between molecular configuration and the optical property is detected. One-photon absorption (OPA), and one-photon emission (OPE), two-photon absorption (TPA), and non-radiative transition are discussed. It is easy to find that NAPBr-m (meta substitution) has a decreased geometry changes, this is responsible for its larger fluorescence quantum efficiency. Through analyzing the electron-hole distribution and transfer properties, the inner mechanism is revealed. We hope our study could provide a reference for designing efficient TPA fluorescent dyes.
In general, oscillator strength is used to characterize the transition probability between the ground state |i⟩ and the excited state |f⟩ molecules in one-photon absorption and emission, which is defined as:
| δop=2ωf3∑α|⟨i|μα|f⟩|2 | (1) |
where ωf denotes excitation energy of the excited state |f⟩, μα is the electric transition dipole moment and the summation is performed over x,y,z axes [10].
The root-mean-square deviation (RMSD), which could be used to measure molecular configuration difference:
| RMSD=√1Nnatom∑i[(xi−x′i)2+(yi−y′i)2+(zi−z′i)2] | (2) |
N represents the number of atoms.
Moreover, the macroscopic TPA cross sections can be defined as [11, 12]:
| σtpa=4π2a05α15c×ω2g(ω)Γfδtpa | (3) |
where a0, α and c are the Bohr radius, the fine structure constant and the speed of light, respectively. g(ω) denotes the spectral line profile, which is assumed to be a δ function here. The level broadening Γf of the final state is commonly assumed to be 0.1 eV, corresponding to a lifetime of about 6 fs [13-15]. The unit of TPA cross sections is GM (1 GM=10−50 cm4⋅s⋅photon−1). Furthermore, the calculation of the fluorescence radiative decay rate can be calculated by Einstein's spontaneous emission equation:
| kr=δΔEfi21.499 | (4) |
where δ is the oscillator strength obtained by optimizing the excited state, ΔEfi2 is the square of vertical emission energy of the excited state with the unit of cm−1. The kr is the radiative decay rate. Besides, based on the Fermi Golden Rule, the non-radiative decay rate calculation formula is
| knr=2πℏ2∑u,vPiv|Hfv,iv|2δ(Eiv−Efu) | (5) |
|Hfv,iv| represents the interaction between two states, knr can be calculated in MOMAP developed by the Shuai group [16-20]. Thus, the fluorescence quantum efficiency ΦPF can be calculated by the following equation:
| ΦPF=krkr+knr | (6) |
We design a series of NAPBr molecules with different substitution sites. The ground state geometry, one-photon absorption and one-photon emission properties are studied. All calculations are performed based on the density functional theory (DFT), using the B3LYP functional and 6-31G(d) basis with the Gaussian 16 program package [21]. Then, the frequency calculations are performed to ensure the stability of the ground state and excited state geometry structures. Considering the solvent effect, the polarizable continuum model (PCM) is adopted for water, CH3CH2OH, and acetone [22]. We evaluate the electron-hole distribution and frontier molecular orbital (FMO) levels. The intramolecular interaction analysis is performed by Multiwfn and rendered in VMD [23, 24]. The RMSD is used to compare the molecular geometry difference of S0 and S1 in VMD. Moreover, the non-radiative decay rate, reorganization energy, and Huang-Rhys (HR) factor are all calculated by MOMAP. The two-photon absorption cross sections are calculated at the B3LYP/6-311G(d) level by Dalton 2013 program. The normal mode displacement and the Duschinsky rotation effect are not considered in this work [25, 26].
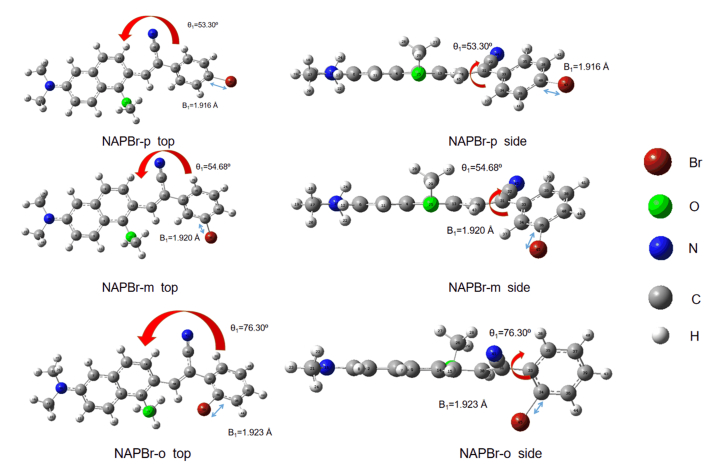
Based on the synthesized molecule in the experiment performed by Tang's group, new dyes are designed to study the effect of substitution sites on the molecular property [2]. Three molecules are named as NAPBr-p/m/o (para/meta/ortho) with different bromine substitution sites are studied in water. Structural formulas of the studied compounds are depicted in FIG. 1. The optimized ground state structures and detailed data are shown in FIG. 2.
In this subsection, the effect of substitution sites on molecular geometrical structures is studied. The dihedral angle θ1 (C14-C13-C33-C35) between benzene ring and naphthalene and bond length B1 (Br-C) are chosen to analyze the molecular ground state configurations. The θ1 values for NAPBr-m, NAPBr-p, and NAPBr-o are 53.3∘, 54.68∘, 76.30∘. The B1 is gradually increased. It shows that NAPBr-o has the largest θ1, reaching 76.30∘, nearly 22∘ larger than that of NAPBr-m. The substitution sites lead to a large twist angle between naphthalene and benzene ring, the coplanar molecular structure is destroyed. For the molecule whose substitution is closer to naphthalene, it has a larger twist angle.
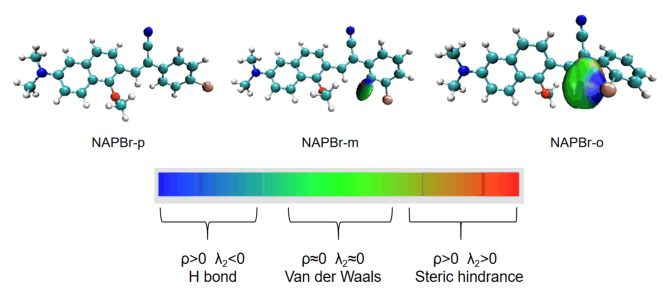
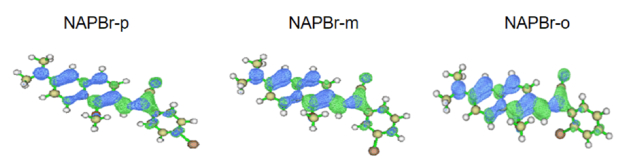
The δg index between atoms is the difference between the two density gradients. It is an important visualization tool to reflect the intramolecular interaction degree between atoms quantitatively. To find out how the substitution site affects benzene ring torsion, we use the independent gradient model (IGM) to draw the δg isosurface diagram. It is effective to show the interaction degree of the bromine atom and the hydrogen atom between benzene ring and naphthalene, which is shown in FIG. 3 [27]. The color change represents the strength and type of the weak interaction. This depends on the value of ρ(r) and λ2. ρ(r) represents the electron density. It can only reflect interaction strength, while the type needs to be reflected by the sign (λ2) function [28, 29]. In FIG. 3, when the red gradually deepens, it represents the deepening of Steric hindrance (ρ < 0 and λ2>0). When the blue gradually deepens, it represents the formation of hydrogen bonds (ρ>0 and λ2<0). In the middle green area, the weak interaction type is van der Waals (ρ≈0 and λ2≈0). The isosurface δg is set to 0.001, so the weak interaction can be displayed well. Moreover, it is selected to obtain a smooth isosurface diagram for the convenience of visualization. It is found that the interaction degree of NAPBr-o is the largest. Isosurface around the bromine atom is blue, so we can recognize that a hydrogen bond is formed. The blue region (large ρ(r) and λ < 0) shows a visible and attractive weak interaction. It further proves the existence of hydrogen bonds [13]. There is a large green area in the adjacent area which shows the electron density in a corresponding weak interaction area is very small and close to zero, so it should be interpreted as dispersion effect [13]. NAPBr-o has a stronger interaction with the hydrogen atom, this is the reason that NAPBr-o has larger benzene ring torsion. Considering the hydrogen bonding interaction, molecular electron distribution change may lead to optical properties differences. It can explain the photophysical property of NAPBr-o differs relatively from the other molecules. More content will be analyzed below.
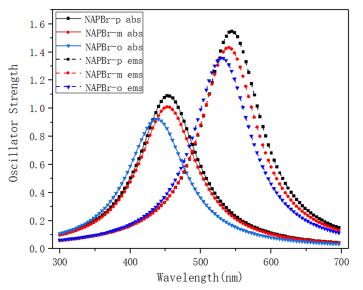
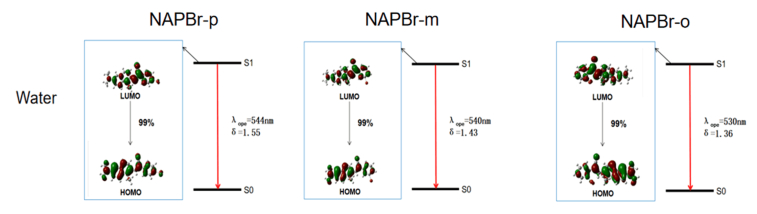
Effective absorption and emission abilities are important embodiments of the optical properties of dye. From FIG. 4, the one-photon absorption (OPA) and one-photon emission (OPE) properties are studied and compared with the experiments. It is easy to find that the absorption wavelengths of NAPBr-p, NAPBr-m, NAPBr-o are 454 nm, 453 nm, 438 nm, respectively. The emission wavelengths are 544 nm, 540 nm, 530 nm, they are sequentially declined. NAPBr-p has the largest emission and absorption wavelengths, corresponding data are collected in Table I. The experimental data collected in THF and the calculated result of the emission wavelengths of NAPBr-p are 525 and 525 nm, and the absorption wavelengths are 409 and 451 nm. The different excitation wavelength, the interaction between the laser and molecules, the inter-molecular interaction including solute-solute interaction result in the discrepancy and they have not been considered in the calculation [28, 29]. The absorption and emission wavelengths of the other molecules show blue shift to NAPBr-p, they have Stokes shift up to 108696 cm−1. Compared with NAPBr-m, the absorption and emission wavelengths of NAPBr-o have a greater blue shift. Eflu is the energy difference between the two electronic states. δabs and δems are the absorption and emission oscillator strength, they are important physical parameters that measure the absorption or emission ability. The absorption and emission oscillator strength are gradually increased. They are related to the fact that molecules have the largest energy gap of HUMO-LUMO (HOMO represents the highest occupied molecular orbital and LUMO represents the lowest unoccupied molecular orbital). Besides, the electric transition dipole moment is also an important factor. Now, detailed analysis for the OPA and OPE processes and transition natures are made to show the optical properties intuitively [28].

|
 DownLoad:
CSV
DownLoad:
CSV
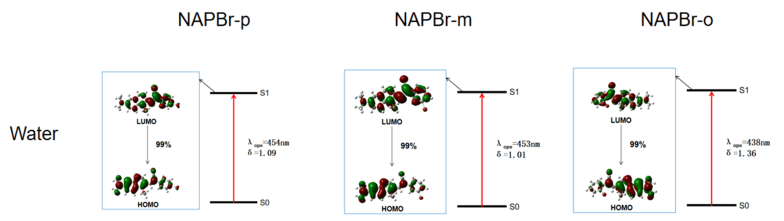
First of all, the OPA processes are given in FIG. 5. It shows that the maximum OPA transition probability state is always the S1. 99% of molecular orbitals are originated from HOMO to LUMO transition. In the molecular absorption process, the molecular orbitals are mainly concentrated in naphthalene which is the fluorescent group. The OPA processes of three molecules are similar.
Then, the OPE processes shown in FIG. 6 are studied. According to Kasha's rule, fast non-radiative internal conversion and vibrational relaxation processes lead to the fact that molecules are located at the lowest vibrational level on S1 after excitation. It also influences the molecular configuration. The analysis is based on the optimized molecules in S0 and S1. It can be seen that S1→S0 transition probability reaches up to 99% from the LUMO to the HOMO transition. It can represent the electron transfer process between fluorescence group and acceptor. Through influencing the orbital distribution overlaps, different substitution sites bring different molecular orbitals transitions. It is the reason that molecular optical property has a regular rule. It accords well with the calculations.
Define the D index that measures the distance between hole and electron centroid [28]:
| Dx=|Xele−Xhole| | (7) |
| Dy=|Yele−Yhole| | (8) |
| Dz=|Zele−Zhole| | (9) |
| Dindex=√(Dx2)+(Dy2)+(Dz2) | (10) |
here, Xele/hole refers to the x coordinate of the holes or electrons mass center. Sr represents the overlap between hole and electron:
| Sr(r)=√ρhole(r)ρele(r) | (11) |
Δσindex reflects the difference in the overall spatial distribution of electrons and holes:
| Δσindex=|σele|−|σhol| | (12) |
We draw molecular holes and electrons distribution diagram to illustrate the electron excitation characteristic in FIG. 7 [30]. The main excitation process is from S0 to S1. The holes and electrons are almost distributed in the naphthalene plane. There is no obvious difference in the holes and electrons distribution. Electrons converge near the torsional benzene ring, while holes are away from the benzene ring. The detailed data are listed in Table II. A large Dindex means that the distance between electrons and holes is large, Sr is large and Δσ is small. It indicates that the overlap between holes and electrons is large. The separation of electrons and holes is not sufficient and the difference in overall spatial distribution is small. It can be considered as charge transfer excitation. Otherwise, Δσ changes regularly. NAPBr-m has the largest Dindex and the smallest Sr, they are related to the distribution and transfer of electrons and holes. It may be closely related to the non-radiative transition, especially the largest fluorescence quantum efficiency.

|
DownLoad:
CSV
A promising candidate for a fluorescent dye should possess a considerable two-photon absorption (TPA) cross section. Therefore, the TPA properties of dyes (such as excitation energy, corresponding TPA cross section, and wavelength) are calculated and compared, corresponding data are presented in Table III. According to the previous investigations, the transition probability of the high-excitation state is small and the transition probability of the low-excitation state (n < 6) is high [31, 32]. Thus, the lowest five excited states are selected in this work. Moreover, the balance between the efficiency and time can be regulated. Thus, the lowest five excited states are selected for analysis in this work.

|
DownLoad:
CSV
Results show that the maximum TPA cross sections of NAPBr-p, NAPBr-m, NAPBr-o are 3658, 2583, and 1490 GM. They are located at the wavelengths of 658, 644, and 633 nm. They are all located at the lowest fourth excited state. Compared with NAPBr-p, the absorption wavelengths of other molecules have a slight blue shift. NAPBr-p has a larger TPA cross section, larger OPA wavelength and OPA oscillator strength. Therefore, it has a better absorption capacity. A good conjugated planarity system and strong acceptor group are important for its excellent molecular absorption ability in Tang's group analysis [2]. The molecular TPA properties trend is consistent with that of OPA. The electric transition dipole moment (μ) plays an important role in affecting TPA property. Electric transition dipole moments are 11.18, 9.52, and 8.85 Debye, respectively. The electric transition dipole moment of NAPBr-p is the largest and NAPBr-o is the smallest. They are in good agreement with TPA property. Besides, the electric transition dipole moments between S0 and S1 are proportional to the orbital overlap between the HOMO and the LUMO. A large electric transition dipole moment presents a large orbital overlap degree and oscillator strength. It can be seen clearly that the electric transition dipole moment, the TPA property, and OPA oscillator strength match well with above mentioned analyses.
Fluorescence quantum efficiency is an important index for evaluating dyes properties. According to Kasha's law, the radiation competition from S1 to S0 has an important impact on fluorescence quantum efficiency, as well as the adiabatic excitation energy. It is easy to see that the radiative decay rates of NAPBr-p, NAPBr-m, NAPBr-o are 3.50×108 s−1, 3.28×108 s−1, and 3.22×108 s−1, respectively. From Table IV, it can be seen that fluorescence quantum efficiency order is NAPBr-m (63.77%)>NAPBr-p (60.56%)>NAPBr-o (52.04%). It is worth mentioning that the fluorescence quantum efficiency of NAPBr-m is the highest. Its radiative decay rate (3.28×108 s−1) is smaller than that of NAPBr-p, but it has the lowest non-radiative decay rate (1.87×108 s−1). Furthermore, the electron-hole distribution and transfer analysis also help one to understand the high fluorescence quantum efficiency of NAPBr-m. Fluorescence quantum efficiency is closely related to the substitution site. The meta-substituted molecule has a stronger ability to suppress the non-radiative loss of excited-state energy, thereby improving the fluorescence quantum efficiency. Combined with the TPA capacities analyzed before, the meta-substituted molecules are ideal dyes. After considering all aspects of performance, we think that NAPBr-m has a better two-photon fluorescent property; it has a larger TPA cross section and the difference of fluorescence quantum efficiency and Stokes shift are smaller than others. The intrinsic mechanism of fluorescence quantum efficiency will be elaborated in the following subsection.

|
DownLoad:
CSV
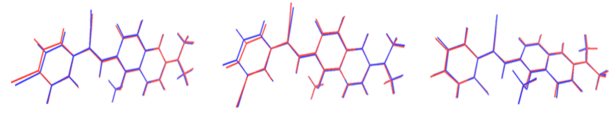
In this part, the influence of substitution sites in the excited state, especially the first excited state is studied in detail. It is very important for fluorescence quantum efficiency. Firstly, to illustrate the effects on the molecular configuration difference of the ground state (S0) and the first excited state (S1) clearly, the root-mean-square deviation (RMSD) is calculated and compared, as shown in FIG. 8. The RMSD values of NAPBr-p, NAPBr-m, and NAPBr-o are 0.230 Å, 0.225 Å, 0.281 Å, respectively. NAPBr-m has the smallest RMSD and NAPBr-o has the largest one. Small RMSD represents that the molecular configuration difference between S0 and S1 is small. The molecular geometric difference mostly comes from benzene ring torsions. It has a close relationship with photo-physical property.
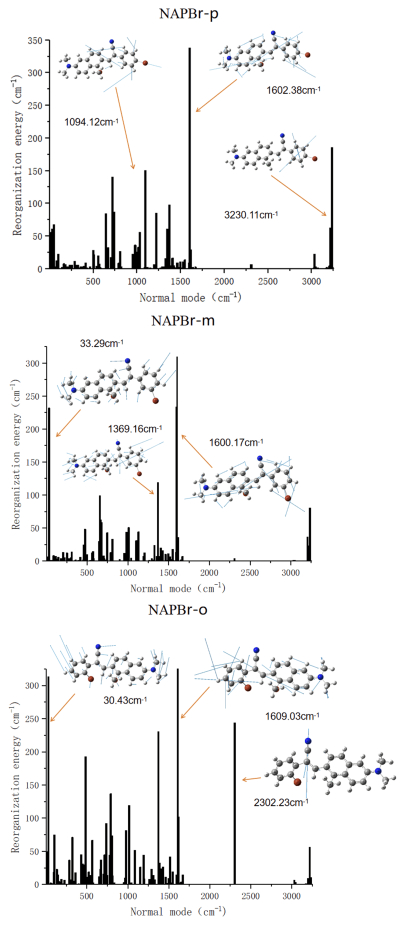
Generally, non-radiative attenuation mainly refers to internal conversion and intersystem crossing. For flexible organic molecules without heavy atoms, the non-radiative attenuation is mainly internal conversion. The non-radiative decay rate is an important index for non-radiative transition. The influence of substituent position on non-radiative transition is analyzed qualitatively in terms of structural change and limited energy loss. And the non-radiative decay rate is related to the geometry changes between the two electronic states. The calculated non-radiative decay rates of NAPBr-p, NAPBr-m, NAPBr-o are 2.28×108 s−1, 1.87×108 s−1, and 2.97×108 s−1, respectively. NAPBr-o has a maximum non-radiative decay rate and a minimum radiative decay rate, thus, the lowest fluorescence quantum efficiency is confirmed. The non-radiative decay rate is related to the energy loss caused by geometric distortion. Moreover, reorganization energy can characterize geometric relaxation degree and internal conversion energy decay effectively, which is shown in FIG. 9. It shows that reorganization energy distributes both in low and high-frequency regions. The order in the low-frequency mode is NAPBr-o, NAPBr-p, NAPBr-m. It is as same as the magnitude reorganization energy order (they are 6808.36 cm−1, 4139.93 cm−1, and 4987.02 cm−1, respectively). The largest value in the high-frequency mode is NAPBr-p, NAPBr-m, NAPBr-o. They are gradually reduced. All molecules have a high-frequency vibration mode at 1600-1610 cm−1 with large reorganization energy (The NAPBr-o reaches 375.23 cm−1). It is mainly caused by the strong vibration of the C=C bond between naphthalene and benzene ring. The direction and magnitude of the arrow on each atom characterize the vibration direction and intensity at this frequency, more data are shown in Table IV. Structural change, especially benzene ring rotation, contributes to molecular large reorganization energy in low-frequency regions. The difference between molecules reflects mainly in the dihedral angle θ1 caused by different substitution sites. It is closely related to the geometric structure difference between the S0 and S1 in the non-radiative transition. The small reorganization energy corresponds to a small non-radiative decay rate. It indicates that the benzene ring torsion is suppressed, resulting in the blocked non-radiative energy loss path [33]. It can also be used to explain the rule of fluorescence quantum efficiency. According to Marcus theory, the electron transfer reactions without making or breaking bonds have smaller geometry relaxation, lower reorganization energy, and higher electron transfer rate. If a molecule has a small geometric difference, it can carry out electron transfer with little vibrating. The consequence is the charges rearrangement. Moreover, NAP AIEgens has a lower TPA cross section (45-100 GM at 860 nm) in the experiment. The fluorescence quantum yield of NAPBr-p is only 1.4% [2]. We believe the solute-molecule or molecule-molecule interaction in the actual environment is the main reason, this issue is not considered in this work.
Then, the Huang-Rhys (HR) factor is calculated to measure the non-radiative excited state energy consumption, characterize the electron vibration coupling, and the contribution degree of geometric configuration difference at this frequency. It helps us further analyze the vibration situation. Some vibration modes, which have larger HR factor (>0.1), are listed in Table V [36]. The Huang-Rhys (HR) factor is calculated by the following equation:
| HRk=ωkDk22 | (13) |

|
DownLoad:
CSV
here ωk represents the vibration frequency and Dk is the normal coordinate displacement of mode k. Combined with the change of θ1, it concludes that larger HR factor is localized in the low-frequency region (<500 cm−1). The contribution degree of structural change is different. Larger HR factor is related to larger benzene ring torsional motion. It can influence the non-radiative decay rate. Small molecular geometry difference between S0 and S1 indicates that NAPBr-m has a small HR factor and high fluorescence quantum efficiency. The benzene rings of NAPBr-p and NAPBr-o twist larger in the low-frequency frequency regions, this brings larger non-radiative decay rate and total HR factor; so, they have lower fluorescence quantum efficiency than NAPBr-m. Thus, the substitution site effects on optical properties are highlighted.
To investigate the solvent effect on optical properties of the studied dyes, the optical properties of three molecules in CH3CH2OH and acetone are calculated. Corresponding data are collected in Table VI. Results show that the substitution site affects molecular TPA ability. NAPBr-p has the maximum TPA cross section that reaches up to 3461 GM and 3375 GM in CH3CH2OH and acetone, respectively. They both have good TPA properties and this feature corresponds well with the electric transition dipole moment. It has been proven that they are promising TPA molecules.

|
DownLoad:
CSV
As we can see from Table VII, NAPBr-o has the maximum non-radiative decay rate and the minimum radiative decay rate. So, its fluorescence quantum efficiency is always the lowest. NAPBr-m has the highest fluorescence quantum efficiency and minimum non-radiative decay rate. It shows the effect of substitution sites on the fluorescence emission properties. Compared with other vibration modes, the reorganization energy and HR factor (< 0.1) are larger in the low-frequency mode (< 500 cm−1), they can be attributed to the benzene ring torsion. Geometric configuration differences lead to different non-radiative energy loss paths. Small geometric configuration difference between S0 and S1 of NAPBr-m makes the non-radiative energy loss path being blocked. So, the non-radiative decay rate is small. Thereby, it has small reorganization energy and total HR factor. This is important for its large fluorescence quantum efficiency. In a general comparison, similar rules are found in three solvents (water, CH3CH2OH, and acetone).

|
DownLoad:
CSV
In this work, we designed and studied a series of new lipid droplets detection dyes based on the experimental dye. They have excellent and regular photophysical properties. Large Stokes shift, remarkable TPA cross section and high fluorescent efficiency are confirmed. Moreover, the studied dyes are proven to have similar rules in different solvents and its inherent mechanism is revealed based on DFT and TD-DFT methods. The influence of different substitution sites on the molecular structures and optics properties are analyzed, transitions from HOMO to LUMO dominate the OPA and OPE processes. The electron-hole distribution and transfer analysis prove that the fluorescent emission mechanism is related to the charge transfer between donor and acceptor. Structural changes (especially benzene ring rotation) play an important role in the non-radiative energy consumption process, the calculated reorganization energy and HR factor in low-frequency regions account for the main part. Furthermore, the influence of different substitution site on fluorescence quantum efficiency is illustrated. The meta-substituted molecule has the stronger ability to suppress non-radiative energy consumption, thereby improving the fluorescence quantum efficiency. After considering all aspects of performance, NAPBr-m has better two-photon fluorescent properties than NAPBr-o and NAPBr-p, and it has the potential to become excellent fluorescent dyes in cell imaging etc.
This work was supported by the National Natural Science Foundation of China (No.11804196 and No.11904210), the Project funded by China Postdoctoral Science Foundation (No.2018M642689), and the Open Fund of Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, (South China University of Technology) (No.2019B030301003).
| [1] |
T. C. Walther and R. V. Farese Jr., Annu. Rev. Biochem. 81, 687 (2012). doi: 10.1146/annurev-biochem-061009-102430
|
| [2] |
G. L. Niu, R. Y. Zhang, J. P. C. Kwong, J. W. Y. Lam, C. P. Chen, J. G. Wang, Y. C. Chen, X. Feng, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams, M. R. J. Elsegood, J. N. Qu, C. Ma, K. S. Wong, X. Q. Yu, and B. Z. Tang, Chem. Mater. 30, 4778 (2018). doi: 10.1021/acs.chemmater.8b01943
|
| [3] |
P. T. Bozza and J. P. B. Viola, Prostaglandins Leukot. Essent. Fatty Acids 82, 243 (2010). doi: 10.1016/j.plefa.2010.02.005
|
| [4] |
A. S. Greenberg, R. A. Coleman, F. B. Kraemer, J. L. McManaman, M. S. Obin, V. Puri, Q. W. Yan, H. Miyoshi, and D. G. Mashek, J. Clin. Invest. 121, 2102 (2011). doi: 10.1172/JCI46069
|
| [5] |
R. C. N. Melo and A. M. Dvorak, PLOS Pathog. 8, e1002729 (2012). doi: 10.1371/journal.ppat.1002729
|
| [6] |
N. Krahmer, R. V. Farese Jr., and T. C. Walther, EMBO Mol. Med. 5, 973 (2013). doi: 10.1002/emmm.201100671
|
| [7] |
D. Kim, H. G. Ryu, and K. H. Ahn, Org. Biomol. Chem. 12, 4550 (2014). doi: 10.1039/C4OB00431K
|
| [8] |
S. Yao and K. D. Belfield, Eur. J. Org. Chem. 2012, 3199 (2012). doi: 10.1002/ejoc.201200281
|
| [9] |
H. M. Kim and B. R. Cho, Chem. Rev. 115, 5014 (2015). doi: 10.1021/cr5004425
|
| [10] |
R. C. Hilborn, Am. J. Phys. 50, 982 (1982). doi: 10.1119/1.12937
|
| [11] |
Y. Luo, P. Norman, P. Macak, and H. Ågren, J. Phys. Chem. A 104, 4718 (2000). doi: 10.1021/jp993803l
|
| [12] |
C. K. Wang, K. Zhao, Y. Su, Y. Ren, X. Zhao, and Y. Luo, J. Chem. Phys. 119, 1208 (2003). doi: 10.1063/1.1579680
|
| [13] |
Y. J. Zhang, J. C. Leng, and W. Hu, Sensors 18, 1324 (2018).
|
| [14] |
Y. J. Zhang, X. Wang, Y. Zhou, and C. K. Wang, Chem. Phys. Lett. 658, 125 (2016). doi: 10.1016/j.cplett.2016.06.026
|
| [15] |
M. Albota, D. Beljonne, J. L. Brédas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. R?ckel, M. Rumi, G. Subramaniam, W. W. Webb, X. L. Wu, and C. Xu, Science 281, 1653 (1998). doi: 10.1126/science.281.5383.1653
|
| [16] |
Y. L. Niu, W. Q. Li, Q. Peng, H. Geng, Y. P. Yi, L. J. Wang, G. J. Nan, D. Wang, and Z. G. Shuai, Mol. Phys. 116, 1078 (2018). doi: 10.1080/00268976.2017.1402966
|
| [17] |
Q. Peng, Y. P. Yi, Z. G. Shuai, and J. J. Shao, J. Am. Chem. Soc. 129, 9333 (2007). doi: 10.1021/ja067946e
|
| [18] |
Y. L. Niu, Q. Peng, and Z. G. Shuai, Sci. China Ser. B Chen. 51, 1153 (2008). doi: 10.1007/s11426-008-0130-4
|
| [19] |
Z. G. Shuai and Q. Peng, Nat. Sci. Rev. 4, 224 (2017). doi: 10.1093/nsr/nww024
|
| [20] |
Z. G. Shuai and Q. Peng, Phys. Rep. 537, 123 (2014). doi: 10.1016/j.physrep.2013.12.002
|
| [21] |
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision B. 01, Wallingford, CT: Gaussian Inc., (2016).
|
| [22] |
J. Tomasi, B. Mennucci, and R. Cammi, Chem. Rev. 105, 2999 (2005). doi: 10.1021/cr9904009
|
| [23] |
T. Lu and F. W. Chen, J. Comput. Chem. 33, 580 (2012). doi: 10.1002/jcc.22885
|
| [24] |
W. Humphrey, A. Dalke, and K. Schulten, J. Mol. Graphics 14, 33 (1996). doi: 10.1016/0263-7855(96)00018-5
|
| [25] |
K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I. M. Høyvik, M. F. Iozzi, B. Jansik, H. J. Aa. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. Rybkin, P. Salek, C. C. M. Samson, A. Sánchez de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski, and H. Ågren, WIREs Comput. Mol. Sci. 4, 269 (2014). doi: 10.1002/wcms.1172
|
| [26] |
Q. Peng, Y. Yi, Z. Shuai, and J. Shao, J. Am. Chem. Soc. 129, 9333 (2007) doi: 10.1021/ja067946e
|
| [27] |
C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras-García, and E. Hénon, Phys. Chem. Chem. Phys. 19, 17928 (2017). doi: 10.1039/C7CP02110K
|
| [28] |
T. Lu, Multiwfn manual, version 3.7(dev), available at: http://sobereva.com/multiwfn (accessed on May 20, 2020).
|
| [29] |
Y. J. Zhang, X. Wang, Y. Zhou, and C. K. Wang, Chem. Phys. Lett. 658, 125 (2016). doi: 10.1016/j.cplett.2016.06.026
|
| [30] |
W. J. Yang, Y. J. Zhang, and C. K. Wang, Acta Phys. Chim. Sin. 31, 2303 (2015). doi: 10.3866/PKU.WHXB201510233
|
| [31] |
Y. Zhou, Y. K. Wang, X. F. Wang, Y. J. Zhang, and C. K. Wang, Chin. Phys. B 26, 083102 (2017). doi: 10.1088/1674-1056/26/8/083102
|
| [32] |
J. Z. Fan, Y. C. Zhang, Y. Zhou, L. L. Lin, and C. K. Wang, J. Phys. Chem. C 122, 2358 (2018). doi: 10.1021/acs.jpcc.7b10238
|
| [33] |
W. L. Cai, H. Z. Zhang, X. Yan, A. C. Zhao, R. X. He, M. Li, Q. X. Meng, and W. Shen, Phys. Chem. Chem. Phys. 21, 8073 (2019). doi: 10.1039/C9CP00819E
|
Figures(9) / Tables(7)

CJCP WeChat
Copyright© 2016 CPS
Address: No.96, JinZhai Road, Hefei, Anhui, 230026, P.R.China
Tel: 0551-63601122 E-mail: cjcp@ustc.edu.cn
Supported by:
Beijing Renhe Information Technology Co., Ltd.

|
|
DownLoad:
CSV
|
|
DownLoad:
CSV
|
|
DownLoad:
CSV
|
|
DownLoad:
CSV
|
|
DownLoad:
CSV
|
|
DownLoad:
CSV
|
|
DownLoad:
CSV